Cohesin gene mutations in tumorigenesis
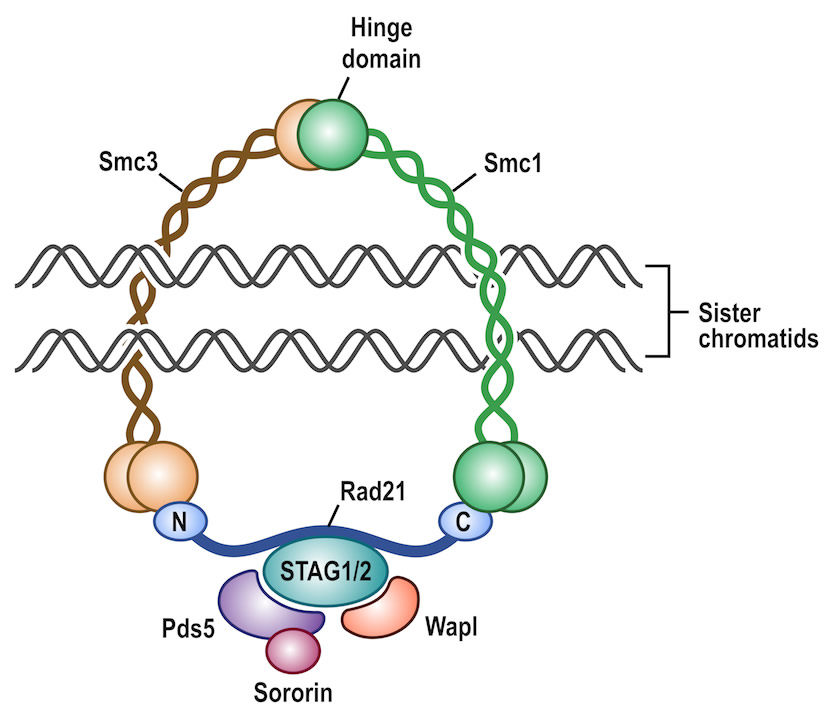
We recently discovered frequent inactivating mutations of genes including STAG2 that encode the multi- protein cohesin complex in several tumors including glioblastoma, urothelial carcinoma, Ewing sarcoma, and acute myeloid leukemia (AML), which define molecular subgroups of these tumors with distinct clinical outcomes. The cohesin complex is responsible for sister chromatid cohesion following DNA replication and helps ensure faithful chromosome segregation during mitosis, but has also been implicated in additional cellular processes such as regulation of chromatin architecture and gene transcription. Our studies in glioblastoma demonstrated that STAG2 mutations were a direct cause of chromosomal instability and aneuploidy; however, cohesin gene alterations in urothelial carcinoma and AML have been identified primarily in near-diploid tumors, suggesting alternative mechanisms by which cohesin inactivation drives oncogenesis. Using a newly generated conditional STAG2 knockout mouse and previously generated isogenic sets of STAG2 proficient and deficient cancer cell lines, we are currently working to determine the function of STAG2 in mouse development and tumorigenesis and to identify therapeutic vulnerabilities in the many cancers harboring cohesin gene alterations. This work is supported by DP5OD021403 from the NIH Director’s Common Fund.
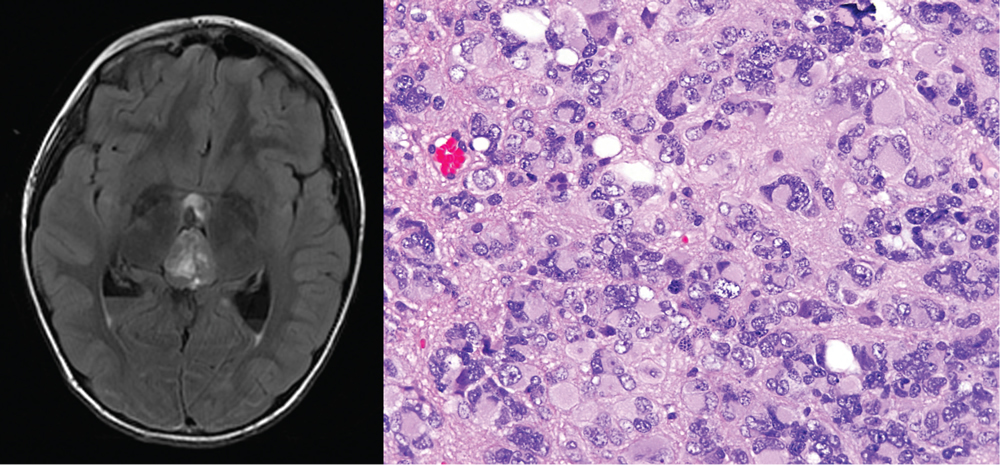
Genomic analysis of pineal parenchymal tumors
The pineal body is a small endocrine gland in the midline of the brain that produces melatonin to modulate circadian rhythms. A group of tumors arise from the pineal gland termed pineal parenchymal tumors which are classified into pineocytomas (grade I), pineal parenchymal tumors of intermediate differentiation (PPTID; grade II or III), and pineoblastomas (grade IV). Typically, pineoblastomas arise in pediatric patients, whereas pineocytomas and PPTIDs occur later in life. Pineocytomas are associated with the most favorable prognosis and rarely disseminate along the craniospinal axis with 5-year survival exceeding 90% following gross total resection. In contrast, pineoblastomas are highly malignant, primitive embryonal tumors with a propensity for cerebrospinal dissemination and poor outcome despite aggressive resection, craniospinal radiation, and systemic chemotherapy. PPTIDs are a morphologically heterogeneous group of tumors with intermediate histologic features and variable clinical outcomes. Rare cases of pineoblastoma have been reported in patients with germline RB1 gene mutation, and more recently a subset of pineoblastomas have been reported in patients with germline DICER1 gene mutations as part of the DICER1 tumor predisposition syndrome. However, the vast majority of pineoblastomas and other pineal parenchymal tumors are sporadic, and the recurrent genetic alterations that drive these pineal parenchymal tumors remain unidentified. We are currently working to characterize the genetic alterations that define each of the pineal parenchymal tumors with the hope of identifying new diagnostic biomarkers and novel therapeutic targets. This work is supported by a Career Development Award from the UCSF Brain Tumor SPORE.
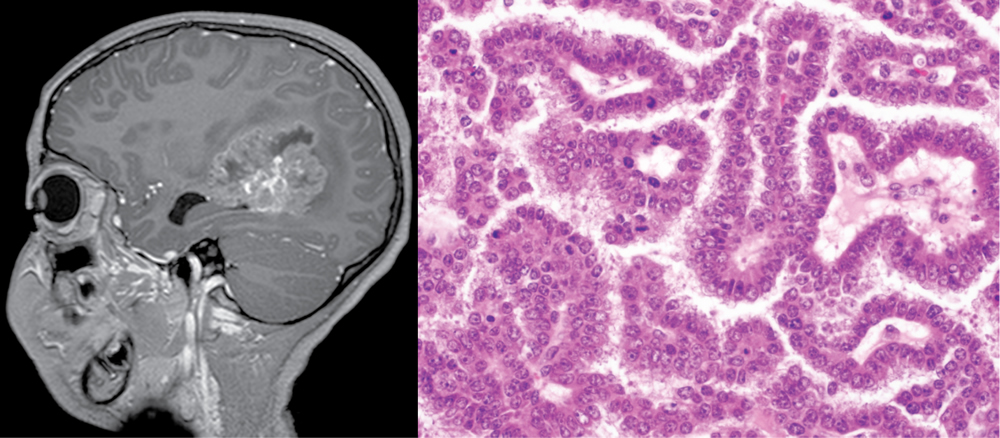
Genomic analysis of choroid plexus tumors
The choroid plexus is an aggregate of modified ependymal cells arranged in papillary cores that reside within each of the ventricles of the central nervous system and is responsible for the production and filtration of the cerebrospinal fluid which bathes the brain and spinal cord. The choroid plexus can give rise to a spectrum of neoplasms ranging from benign papillomas to frankly malignant carcinomas. While choroid plexus tumors are quite rare accounting for approximately 0.5% of all intracranial neoplasms, they account for up to 4% of all brain tumors in children under 15 years of age and up to 20% of those in patients less than 1 year old. Despite aggressive multimodal therapy including adjuvant radiation and chemotherapy, long-term survival remains poor in patients with choroid plexus carcinoma. While a subset of choroid plexus carcinomas occur in patients with germline TP53 mutations as part of Li-Fraumeni syndrome, the majority of both choroid plexus papillomas and carcinomas are sporadic and the genetic alterations that drive these tumors remain unidentified. Our studies aim to delineate the molecular pathways that drive choroid plexus tumors with the potential to improve diagnosis, prognostic classification, and identify new therapeutic targets for this group of poorly understood primary brain tumors. This work is supported by the UCSF 500 Cancer Gene Panel Pilot Program.